Alzheimer's Complexity: Beyond the APOE Gene
This poster was a fun project to raise awareness about Alzheimer’s and highlight that it’s more than just genetics. Written in 2023. Type of Medical writing: research paper. Audience: Undergraduate Life Science Students. Complexity: Level III
Dhruvini
3/4/2026


Introduction
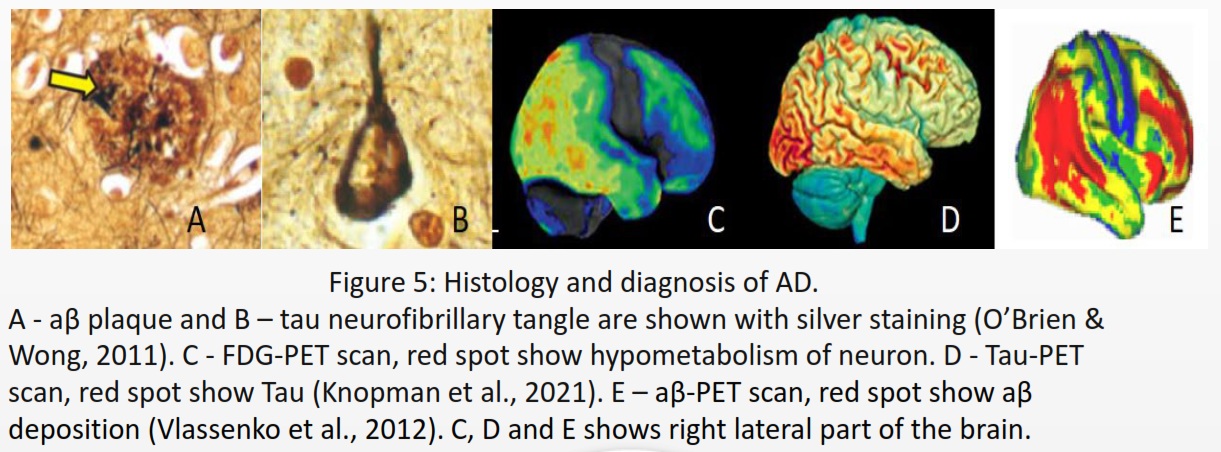
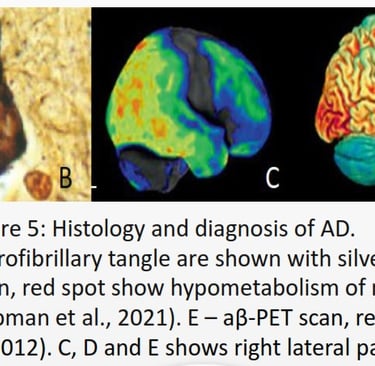
Alzheimer’s disease (AD) is a type of neurogenerative disease and the most common form of dementia. Common affected areas are medial temporal lobe, which includes the hippocampus, amygdala, and Para hippocampal regions (De-Paula et al., 2012), responsible for declarative memory related to facts and events (Squire et al., 2004). It impacts the neocortical structure in the medial prefrontal cortex, which is in charge of cognitive processes and emotional regulation (Xu et al., 2019). According to the NIH National Institute on Aging (NIA), symptoms include misplacing items, forgetting appointments, and communication problems. Signs progress from poor memory or judgment, personality changes to seizures, numbness to the surrounding area, and aspiration pneumonia, leading to loss of organ functionality and death. AD is identified by the presence of neurofibrillary tangles composed of tau located in the cytoplasm and amyloid-β (aβ) plaques commonly found in the extracellular matrix (ECM) of neurons.
Amyloid precursor protein (APP) gene found on 21q21.2-3 has high transcription activity to produce β-amyloid precursor protein (βAPP), a type 1 transmembrane protein receptor for signaling cell survival, migration, or cell growth (Vu Nguyen, 2019). As a regulation or control point, neurons undergo a high degradation rate of βAPP using three pathways: the non-amyloidogenic pathway, the amyloidogenic pathway, and the N-secretase pathway. Amyloidogenic pathway, when β-secretase digests βAPP into soluble ectodomain of APP (sAPPβ) and C-terminal fragment (CTF-β). γ-Secretase digests CTF-β into Aβ peptide and the APP intracellular domain (AICD). AICD binds to Fe65 to transcribe histone acetyltransferase, and TIP60 is used for various gene regulation activities (Coronel et al., 2018).
The aβ are β-pleated proteins that act as monomers stacking on top of each other, forming long fibril polymers (Seeman & Seeman, 2011). It has no physiological purpose The enzymatic pathway involves neprilysin (NEP), insulin-degrading enzyme (IDE), matrix metalloproteinase (MMP)-9, and glutamate carboxypeptidase II (GCPII) to degrade aβ (Numata & Kaplan, 2010). The nonenzymatic pathway includes transferring a large volume of interstitial fluid (ISF) into cerebrospinal fluid (CSF) and back again into ISF, where aβ drains into the circulatory system to be degraded by the liver. aβ undergoes phagocytosis by microglia or astrocytes. aβ binds to low-density lipoprotein receptor-related protein 1 (LRP1), very low density lipoprotein receptor (VLDLR), and P-glycoprotein to be transported across the blood brain barrier (BBB) into the circulatory system (Yoon & AhnJo, 2012). Tau protein is transient between α-helices and β-pleated sheets and encoded by the microtubule-associated protein tau (MAPT) gene located on17q21.31 (Mandelkow & Mandelkow, 2012). Tau is functional during post-translation modification (PTM) by phosphorylation to stabilize microtubules at the distal axon. The pathology of tau arises from misfolding, either from mutation at MAPT/mRNA or error during PTM, that results in a change in 3D dimension resembling a prion that resists ubiquitination and proteolysis (Muralidar et al.,2020).
Aβ and tau directly interact with each other, providing a positive feedback loop. Overtime accumulation of both tau and aβ causes necrosis and apoptosis of neurons, causing activation of glia cells and releasing inflammatory molecules and cytokines, resulting in neuron death. This progresses at the tissue level and eventually develops scars in the brain (Lee & MacLean, 2015).The purpose of the poster is to identify apolipoprotein E (APOE) gene variants within the sample and explore causes for the increased rate of AD shown in figure 3. Hypothesis: lack of APOE4 gene due to no AD observed within four generations of the family.
APOE Gene
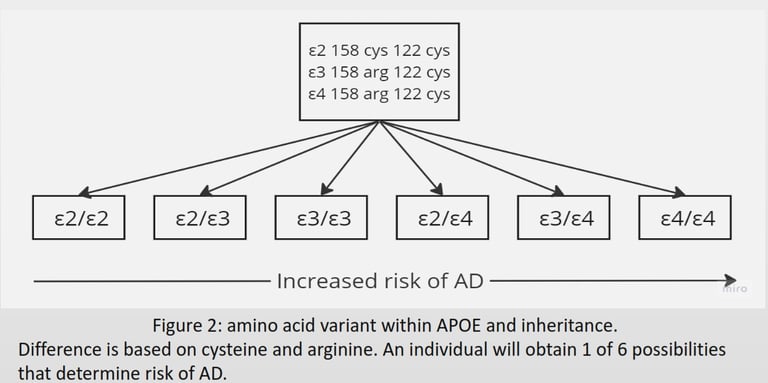
Cholesterol is a vital steroid lipid used for dendrite formation, synapse formation, and synaptic vesicle for neurotransmitter storage. It is synthesised and packaged in lipoprotein for transportation by astrocytes (Orth & Bellosta, 2012). The Apolipoprotein E (APOE) gene on 19q13.32 encodes for the APOE protein used as cell membrane proteins on lipoprotein to bind LDL receptor family to induce endocytosis. The APOE gene is semidominant, expressing three variants (ε2, ε3, and ε4) with six possibilities differing in amino acids (Figure 2) (Lane-Donovan & Herz, 2017). Ε4 is the most vulnerable to degradation; upon cleavage, the fragments form ε4- aβ complex that resists the enzymatic pathway and damages BBB. ε4- aβ downregulates VLDLR and LRP1 increasing aβ providing positive feed back loop for ε4- aβ. (Pires & Rego, 2023). Ε2 is associated with reduced risk due to its ability to upregulate the nonamyloidogenic pathway and VLDLR to reduce aβ deposition (Li et al., 2020).
Diagnosis/Treatment/Epidemiology
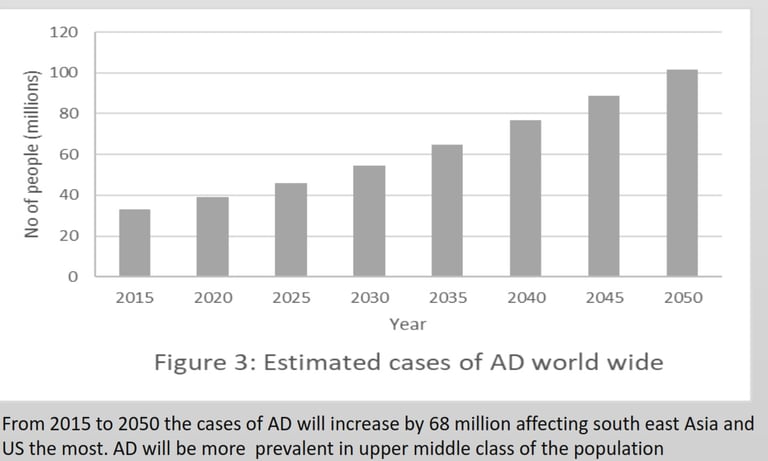
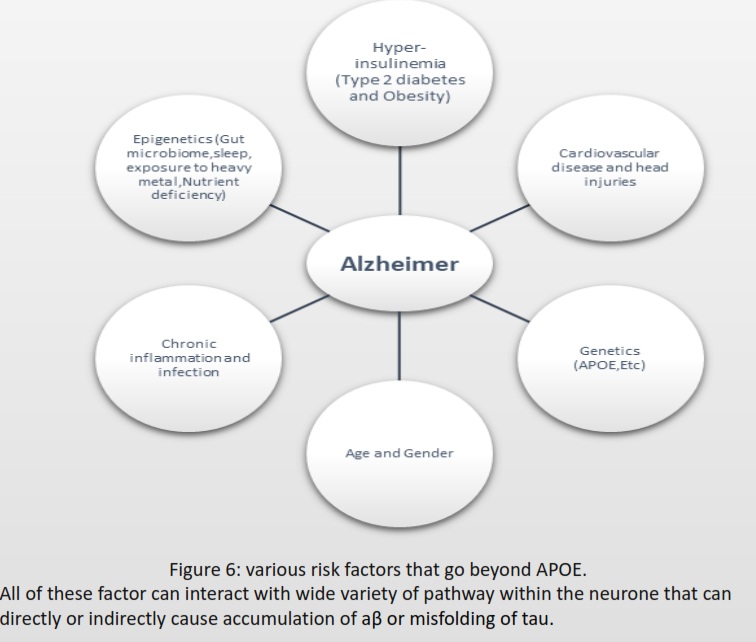
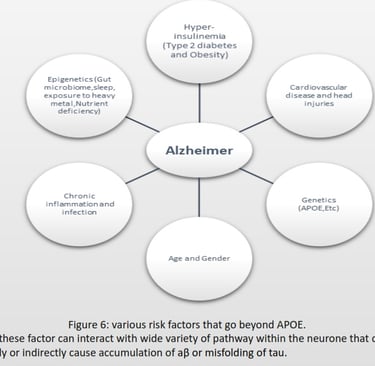
AD cannot spread like an infectious disease, but the progression depends on the risk factors summarized in Figure 6.The first step for diagnosis is a mild cognitive impairment assessment (MCI) to identify memory, planning, judgement, visual, language, and physica impairments. If ≥ 1 is impaired, the individual should proceed to fluorodeoxyglucose positron emission tomography (FDG-PET) to measure neuron metabolism. If positive, using aβ-PET or TauPET, specific for AD should be carried out (Knopman et al., 2021).There is no cure for AD, and treatment focuses on symptoms, including donepezil, galantamine, and rivastigmine, classified as acetylcholinesterase. Inhibitors to inhibit cholinesterase increase acetylcholine for enhanced communication between neurons. AD has an excessive level of glutamate that overexcites neurons. Memantine inhibits the N-methyl-D-aspartate (NMDA) receptor for glutamate to prevent the influx of calcium and sodium ions, reducing the load on neurons (Vaz & Silvestre, 2020).According to Alzheimer’s Disease International (ADI), two-thirds cases of dementia are AD. During 2020, 39 million cases of AD was recorded, and by 2050, it estimate is 101 million worldwide. APOE variant arose 400 million years ago (Huebbe & Rimbach, 2017), Ε4 inheritance is not sufficient to explain the increase of 62 million cases just within 30 years.
Methodology
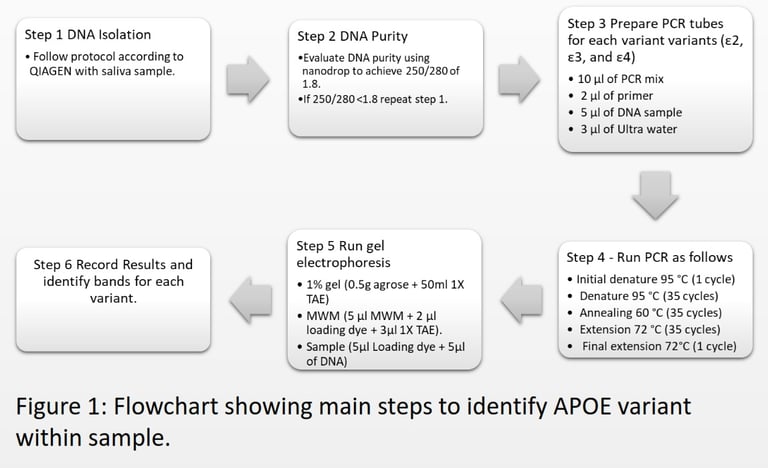
Results
Discussion
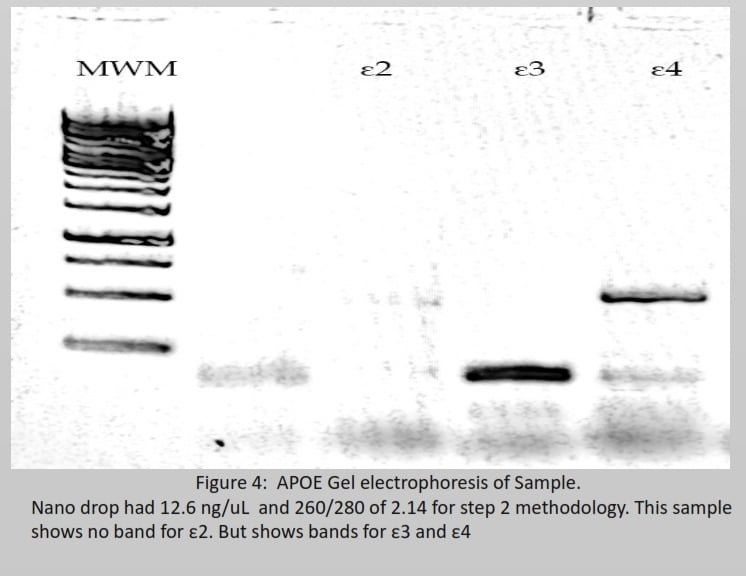
Figure 2 describes six possible inheritances of apoe, arranged from ε2/ε2 being protective against AD to ε4/ε4 being high risk for AD. To determine which variant a sample has, figure 4 shows 2 bands at ε3 and ε4. Both bands should be between 200 bp and 100 bp; this has only happened in ε3. Therefore, the band in ε4 is noise, and the final inheritance for this sample is ε3/ε3 being neural for AD (Pantelidis et al., 2003). Most of the risk factors arise from its ability to directly or indirectly interact with multiple biochemical pathways that lead to upregulation or downregulation of receptors or enzymes, a change in DNA, and the denature or degradation of signalling proteins that have the potential to accumulate aβ and modify tau. AD individuals maintain reduced gut diversity, where pathogenic strains in the gut can release toxins that cross the BBB (Vogt et al., 2017). Vitamin C, folate, vitamin D, and vitamin B12 defiency increases reactive oxygen species (ROS) and advanced glycation end (AGE), which can directly cause DNA mutation or protein denature. Lead and aluminium cause the misfolding of tau. Poor sleep prevents the drainage of ISF during the non-enzymatic pathway. Infections such as the herpes simplex virus (HSV-1), Treponema pallidum, and Chlamydia pneumonia trigger an inflammatory response, generating a high level of cytokines and ROS. Stroke and head injuries can introduce hypoxia within the neuron to increase aβ and tau. Hyperinsulinemia downregulates IDE and increases chronic inflammation, inducing a high level of ROS for aβ and tau accumulation. Menopause causes a reduction in oestrogen, used for cell survival, and a reduction in aβ (Breijyeh & Karaman, 2020). Autophagy activity significantly reduces with age for degradation of damaged organelles and proteins by fusing with selective lysosomes. According to (Hamano et al., 2021), AD requires downregulation of human presenilin 1 (PS1) to prevent lysosomes from degrading tau and aβ for macroautophagy during early events. Autophagic activity can be enhanced by the absence of type 2 diabetes or nutrient deficiency and regular exercise (Barbosa et al., 2019). Risk factors in Figure 6 explain the rise in AD despite ε4 remaining constant within the population for over 100 years.
Conclusion
New therapeutics are based on β-secretase and γ-secretase inhibitors that affect only a few of these interactions and pathways. Other therapies degrade Aβ using antibodies or vaccines may show promising results for slowing down AD (Vaz & Silvestre, 2020). The chances of discovering a cure by 2050 are low. With an increasing population and an increasing cost of healthcare, the most effective strategy to stabilise the graph in Figure 3 is prevention of the disease.
References
1. Barbosa, M. C., Grosso, R. A., & Fader, C. M. (2019). Hallmarks of Aging: An Autophagic Perspective. Frontiers in Endocrinology, 9. https://doi.org/10.3389/fendo.2018.00790
2. Breijyeh, Z., & Karaman, R. (2020). Comprehensive Review on Alzheimer’s Disease: Causesand Treatment. Molecules, 25(24), 5789. https://doi.org/10.3390/molecules25245789
3. Coronel, R., Bernabeu-Zornoza, A., Palmer, C., Muñiz-Moreno, M., Zambrano, A., Cano, E., & Liste, I. (2018). Role of Amyloid Precursor Protein (APP) and Its Derivatives in the Biology andCell Fate Specification of Neural Stem Cells. Molecular Neurobiology, 55(9), 7107–7117. https://doi.org/10.1007/s12035-018-0914-2
4. De-Paula, V. J., Radanovic, M., Diniz, B. S., & Forlenza, O. V. (2012). Alzheimer’s Disease. Protein Aggregation and Fibrillogenesis in Cerebral and Systemic Amyloid Disease, 65, 329– 352. https://doi.org/10.1007/978-94-007-5416-4_14
5. Hamano, T., Enomoto, S., Shirafuji, N., Ikawa, M., Yamamura, O., Yen, S.-H., & Nakamoto, Y. (2021). Autophagy and Tau Protein. International Journal of Molecular Sciences, 22(14),7475. https://doi.org/10.3390/ijms22147475
6. Huebbe, P., & Rimbach, G. (2017). Evolution of human apolipoprotein E (APOE) isoforms: Gene structure, protein function and interaction with dietary factors. Ageing Research Reviews, 37, 146–161. https://doi.org/10.1016/j.arr.2017.06.002
7. Knopman, D. S., Amieva, H., Petersen, R. C., Chételat, G., Holtzman, D. M., Hyman, B. T., Nixon, R. A., & Jones, D. T. (2021). Alzheimer disease. Nature Reviews Disease Primers, 7(1). https://doi.org/10.1038/s41572-021-00269-y
8. Lane-Donovan, C., & Herz, J. (2017). ApoE, ApoE receptors, and the Synapse in Alzheimer’s Disease. Trends in Endocrinology and Metabolism: TEM, 28(4), 273–284. https://doi.org/10.1016/j.tem.2016.12.001
9. Lee, K. M., & MacLean, A. G. (2015). New advances on glial activation in health and disease. World Journal of Virology, 4(2), 42–55. https://doi.org/10.5501/wjv.v4.i2.42
10.Li, Z., Shue, F., Zhao, N., Shinohara, M., & Bu, G. (2020). APOE2: protective mechanism and therapeutic implications for Alzheimer’s disease. Molecular Neurodegeneration, 15(1). https://doi.org/10.1186/s13024-020-00413-4
11.Mandelkow, E.-M. ., & Mandelkow, E. (2012). Biochemistry and Cell Biology of Tau Protein in Neurofibrillary Degeneration. Cold Spring Harbor Perspectives in Medicine, 2(7), a006247–a006247. https://doi.org/10.1101/cshperspect.a006247
12.Muralidar, S., Ambi, S. V., Sekaran, S., Thirumalai, D., & Palaniappan, B. (2020). Role of tau protein in Alzheimer’s disease: The prime pathological player. International Journal of Biological Macromolecules, 163, 1599–1617. https://doi.org/10.1016/j.ijbiomac.2020.07.327
13.Numata, K., & Kaplan, D. L. (2010). Mechanisms of Enzymatic Degradation of Amyloid Beta Microfibrils Generating Nanofilaments and Nanospheres Related to Cytotoxicity. Biochemistry, 49(15), 3254–3260. https://doi.org/10.1021/bi902134p
14.O’Brien, R. J., & Wong, P. C. (2011). Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annual Review of Neuroscience, 34(1), 185–204. https://doi.org/10.1146/annurevneuro-061010-113613
15.Orth, M., & Bellosta, S. (2012). Cholesterol: Its Regulation and Role in Central Nervous System Disorders. Cholesterol, 2012, 1–19. https://doi.org/10.1155/2012/292598
16.Pantelidis, P., Lambert-Hammill, M., & Wierzbicki, A. S. (2003). Simple Sequence-specificPrimer-PCR Method To Identify the Three Main Apolipoprotein E Haplotypes. Clinical Chemistry, 49(11), 1945–1948. https://doi.org/10.1373/clinchem.2003.021683
17.Pires, M., & Rego, A. C. (2023). Apoe4 and Alzheimer’s Disease Pathogenesis-Mitochondrial Deregulation and Targeted Therapeutic Strategies. International Journal of Molecular Sciences, 24(1), 778. https://doi.org/10.3390/ijms24010778
18.Seeman, P., & Seeman, N. (2011). Alzheimer’s disease: β-amyloid plaque formation in human brain. Synapse, 65(12), 1289–1297. https://doi.org/10.1002/syn.20957
19.Squire, L. R., Stark, C. E. L., & Clark, R. E. (2004). THE MEDIAL TEMPORAL LOBE. Annual Review of Neuroscience, 27(1), 279–306. https://doi.org/10.1146/annurev.neuro.27.070203.144130
20.Vaz, M., & Silvestre, S. (2020). Alzheimer’s disease: Recent treatment strategies. European Journal of Pharmacology, 887(1), 173554. https://doi.org/10.1016/j.ejphar.2020.173554
21.Vlassenko, A. G., Benzinger, T. L. S., & Morris, J. C. (2012). PET amyloid-beta imaging in preclinical Alzheimer’s disease. Biochimica et Biophysica Acta (BBA) - Molecular Basis of Disease, 1822(3), 370–379. https://doi.org/10.1016/j.bbadis.2011.11.005
22.Vogt, N. M., Kerby, R. L., Dill-McFarland, K. A., Harding, S. J., Merluzzi, A. P., Johnson, S. C., Carlsson, C. M., Asthana, S., Zetterberg, H., Blennow, K., Bendlin, B. B., & Rey, F. E. (2017).Gut microbiome alterations in Alzheimer’s disease. Scientific Reports, 7(1).https://doi.org/10.1038/s41598-017-13601-y
23.Vu Nguyen, K. (2019). β-Amyloid precursor protein (APP) and the human diseases. AIMS Neuroscience, 6(4), 273–281. https://doi.org/10.3934/neuroscience.2019.4.273
24.What Are the Signs of Alzheimer’s Disease? (2022, October 18). National Institute on Aging. https://www.nia.nih.gov/health/alzheimers-symptoms-and-diagnosis/what-are-signsalzheimers-disease
25.Xu, P., Chen, A., Li, Y., Xing, X., & Lu, H. (2019). Medial prefrontal cortex in neurological diseases. Physiological Genomics, 51(9), 432–442.https://doi.org/10.1152/physiolgenomics.00006.2019
26.Yoon, S.-S., & AhnJo, S.-M. (2012). Mechanisms of Amyloid-β Peptide Clearance: Potential Therapeutic Targets for Alzheimer’s Disease. Biomolecules and Therapeutics, 20(3), 245–255. https://doi.org/10.4062/biomolther.2012.20.3.245
